A recent discussion on a paper I’m writing made me take a step back and ponder what do I really know about GLMs? After a few moments I realised expanding the acronym to Generalised Linear Model was about as far as I could get. So decided it was time to crack this nut.
So a GLM is made up of three key components:
- The error structure
- The linear predictor.
- The link function.
A GLM allows models to incorporate a variety of different error distributions, this is really useful when dealing with data that isn’t normally distributed, such as count data (Poisson errors), presence/absence data (binomial errors) which is really useful depending on the data you have and the questions you are intending to ask.
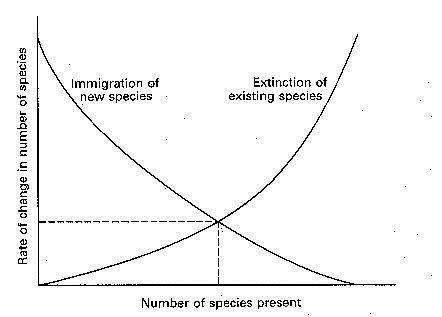
GLM have a number of applications, but one common use is to make predictions on species distributions. GLMs relate an observed value to a predicted value. The observed values are essentially your raw data or say the environmental variables at site x, the predicted values are obtained by transforming the linear predictor. The linear predictor is the sum of the effects of one or more explanatory variables used in the model.
In the model I’ve been using I been mixing GLMs using finite mixture models, I won’t go into detail on mixing GLMs, but in my example the model is comprised of four GLMs, the below R code is a way at looking how well the models are fitting the data. I’ll be showing you how to explore an individual GLM.
An example of generalised linear model equation:
> final.formula <- final.mod$formula
> y ~ t + tsq + s + ssq + o + osq + flux3 + flux3sq
In the above equation y (chance of observing this groups of species) is explained by the environmental covariates t = temperature, s= salinity, o = oxygen concentration and flux3 = particulate organic carbon drift to the sea floor (a measure of productivity) and there squared versions (for looking at non-linear relations of the data).
Note: you can use the poly function in R to fit polynomials to your data,
Eg: poly(t, 2) is the same at tsq above.
In essence the linear predictor is the observed values * the model coefficients. Here is an example of calculating the linear predictors across the sites used to fit the above model.
# This gives you the environmental variables for each site in my model
> model.env.data <- model.matrix(final.formula,data=cbind(obs=1,env_data))
# This gives the linear predictor across all these sites.
> lpre <- model.env.data%*%final.mod$coef[1,]
To determine the fit of a given model, a GLM evaluates the linear predictor for each value of the response variable, then compares the predicted value with a transformed value of y. The transformation to be employed is specified in the link function
The link function can take a number of different forms depending on the type of error structure in the model. Classically there are four main link functions which can be used to transform y. If your data is normally distributed: identity link; poisson: log link; binomial: logit and gamma: reciprocal. In my example I’m using presence/absence data which means I have a binomial error distribution and hence I use a logit link function.
#Setting the link function
> link.fun <- make.link(“logit”)
If you are using a function in R such as glm you can define the link function when you enter the model formula:
> glm(y ~ t + tsq + s + ssq + o + osq + flux3 + flux3sq, family=binomial)
The size of each term in linear predictor indicates it contribution to the overall likelihood, big negative indicates low probability, big positive indicates high probability.
To get the overall log likelihood you transform the linear predictor using the link function as is shown below:
> ll <- link.fun$linkinv(sum(lpre))
However often you want to have a look at how the environmental covariates (coefficients) are responding within the model. There are a number of ways to do this but one way that works quite well is to visualise the response. For instance if you wanted to see how temperature was responding you could look at what range of values temperature contribute positively or negatively to overall log likelihood.
Here is a little bit of code which plots the response of temperature in the above model.
> tmp.temp <- seq(2,26,length=100)
> t.plot <- plot(tmp.temp,final.mod$coef[1,2]*tmp.temp+final.mod$coef[1,3]*tmp.temp^2,type=”l”,ylab=”Total contribution of Temperature to Likelihood in G1″,xlab=”Temperature”)
If everything worked correctly you should a figure such as the one below:

In this figure you can see the non-linear response of temperature and can see for temperatures greater than 20°C temperature is contributing to the overall likelihood of the model. You could potentially do this with all the environmental covariates and visualise their respective responses.
If you want to be a bit fancy you can also add in the intercept:

plot(tmp.temp,final.mod$coef[1,1]+final.mod$coef[1,2]*tmp.temp+final.mod$coef[1,3]*tmp.temp^2,type=”l”,ylab=”Total contribution of Temperature to Likelihood + Intercept”, xlab=”Temperature”)
Another potential use of the linear predictor is to check the contribution of environmental variables when using a model to make predictions across an extent. This problem first cropped up when I was getting extremely high probabilities at sites where it didn’t correlate with other the majority of high probabilities.
To assess the contributions of environmental variables at prediction sites, you can follow a similar pattern to the code above but instead of entering the environmental covariates used to fit the model you enter the environmental variables that you want to predict your model onto.
model.pred.data <- model.matrix(final.formula,data=cbind(obs=1,PrePoint))
link.fun <- make.link(“logit”)
lpre.pred <- model.pred.data%*%final.mod$coef[1,]
## log likelihood
ll <- link.fun$linkinv(lpre.pred)
This will give you the log likelihood for all the points in space. Once you have achieved this you can look at the data points that are producing abnormally high log likelihood values.
> ll.sort <- head(sort(ll, decreasing=TRUE))
An example in my model was site 20087 where the probability of was detection was abnormally high and not aggregated with the majority of high probability points. You can manually look at the environmental covariates by entering the coefficient values from the model and environmental variables from site x (in this case 20087), but keeping all values except one (temperature in this case) at the mean.
Which should look something like this:
lpre.g1.20087.t <- 3522.88236315 + -0.23010015*27.21660 + 0.01096224*tsqmn + -200.04575957*smn + 2.84021577*ssqmn + -1.98680350*omn + 0.36839100*osqmn + -0.43793932*flux3mn + 0.01525628*flux3sqmn
Environmental variables at site 20087:
(Intercept) t tsq s ssq o osq flux3 flux3sq
1.000 27.2166 740.743 34.421 1184.825 4.494 20.197 2.379 5.662
> lpre.g1.20087.t
[1] -5.747775
From this you can see that temperature isn’t contributing to the overall log likelihood at this site. After some detective work I found that salinity was the culprit. See below:
> lpre.g1.20087.s <- 3522.88236315 + -0.23010015*tmn + 0.01096224*tsqmn + -200.04575957*34.4212 + 2.84021577*ssqmn + -1.98680350*omn + 0.36839100*osqmn + -0.43793932*flux3mn + 0.01525628*flux3sqmn
> lpre.g1.20087.s
[1] 79.23035
Going back to those nifty graphs above:
tmp.sal <- seq(34,36,length=100)
> plot(tmp.sal,final.mod$coef[1,4]*tmp.sal+final.mod$coef[1,5]*tmp.sal^2, type=”l”,ylab=”Total contribution of Salinity to Likelihood in G1″,xlab=”Salinity”)

You can see that the values for salinity at 34.4 are contributing highly to the likelihood of the overall model and could account for the really high probability at this site. With some further investigation I found that the environmental covariate values sat outside the bounds of my model space. This is a problematic issue as you can’t estimate a relationship where you don’t have any data. So the best thing to do in this situation is to remove these points from my prediction space. Once I clipped the sites with environmental variables outside the sites used to fit my initial model things started looking good.
Well that’s my first GLM blog. I hope that it makes sense and doesn’t raise to many questions. I’d love some further advice on dealing with GLMs in R so feel free to post a comment or two. Cheers Skip.



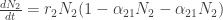
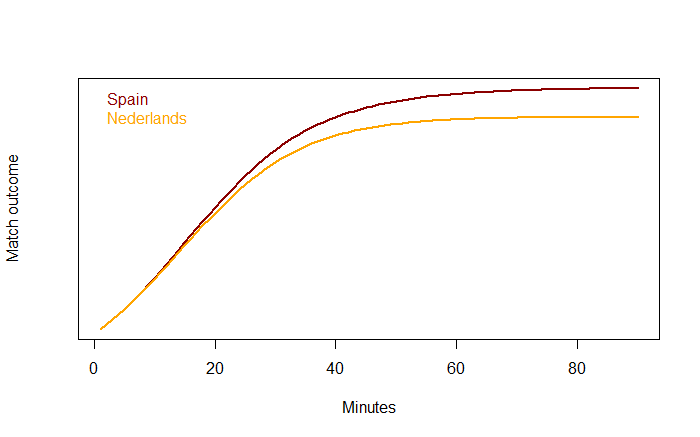
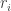 Represents individuals produced per individual per unit time. As we are thinking of this in terms of football, this might be the appearance of new players? However as that doesn’t really make sense in terms of a football match, let’s think of this as number of passes achieved at each time step. A proxy for a teams competitive edge.
Represents individuals produced per individual per unit time. As we are thinking of this in terms of football, this might be the appearance of new players? However as that doesn’t really make sense in terms of a football match, let’s think of this as number of passes achieved at each time step. A proxy for a teams competitive edge.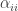 Is the effect of intra-specific competition between individuals of species i on the overall population size. Once again in terms of football this might represent the tempo as game is played at? If the tempo (speed a team plays) is fast, you might expect to out play the opposition. If you play really fast then we might expect teams to get tired, their passing starts to fall apart and eventually you lose due to exhaustion. Additionally, if a team’s tempo is too slow and you lose (Unless you’re the Spanish team, as we’ll discover in the example below).
Is the effect of intra-specific competition between individuals of species i on the overall population size. Once again in terms of football this might represent the tempo as game is played at? If the tempo (speed a team plays) is fast, you might expect to out play the opposition. If you play really fast then we might expect teams to get tired, their passing starts to fall apart and eventually you lose due to exhaustion. Additionally, if a team’s tempo is too slow and you lose (Unless you’re the Spanish team, as we’ll discover in the example below). Is the negative effect of inter-specific competition of species j has on the growth rate of species i or vice versa. For the purposes of my football example, we are going to think of this as pressure from the opposing team.
Is the negative effect of inter-specific competition of species j has on the growth rate of species i or vice versa. For the purposes of my football example, we are going to think of this as pressure from the opposing team.